
| Synonyms | |
| Status | |
| Molecule Category | Free-form |
| UNII | 5DX9U76296 |
| EPA CompTox | DTXSID2046919 |
Structure
| InChI Key | YCOYDOIWSSHVCK-UHFFFAOYSA-N |
|---|---|
| Smiles | |
| InChI |
|
Physicochemical Descriptors
| Property Name | Value |
|---|---|
| Molecular Formula | C20H15ClN4 |
| Molecular Weight | 346.82 |
| AlogP | 5.01 |
| Hydrogen Bond Acceptor | 4.0 |
| Hydrogen Bond Donor | 1.0 |
| Number of Rotational Bond | 4.0 |
| Polar Surface Area | 50.7 |
| Molecular species | NEUTRAL |
| Aromatic Rings | 4.0 |
| Heavy Atoms | 25.0 |
Pharmacology
| Mechanism of Action | Action | Reference |
|---|---|---|
| Macrophage colony stimulating factor receptor inhibitor | INHIBITOR | PubMed PubMed |
| Platelet-derived growth factor receptor inhibitor | INHIBITOR | PubMed PubMed |
| Stem cell growth factor receptor inhibitor | INHIBITOR | PubMed PubMed |
| Vascular endothelial growth factor receptor inhibitor | INHIBITOR | PubMed PubMed |
| Targets | EC50(nM) | IC50(nM) | Kd(nM) | Ki(nM) | Inhibition(%) | |
|---|---|---|---|---|---|---|
|
Enzyme
Kinase
Protein Kinase
Atypical protein kinase group
Atypical protein kinase PIKK family
Atypical protein kinase FRAP subfamily
|
- | - | - | - | 49 | |
|
Enzyme
Kinase
Protein Kinase
TK protein kinase group
Tyrosine protein kinase Abl family
|
- | - | - | - | 25 | |
|
Enzyme
Kinase
Protein Kinase
TK protein kinase group
Tyrosine protein kinase DDR family
|
- | - | 270-270 | - | - | |
|
Enzyme
Kinase
Protein Kinase
TK protein kinase group
Tyrosine protein kinase EGFR family
|
- | 457.7 | - | - | 46-67 | |
|
Enzyme
Kinase
Protein Kinase
TK protein kinase group
Tyrosine protein kinase FGFR family
|
- | - | - | - | 31 | |
|
Enzyme
Kinase
Protein Kinase
TK protein kinase group
Tyrosine protein kinase PDGFR family
|
- | 490-730 | 5.1-700 | - | 72 | |
|
Enzyme
Kinase
Protein Kinase
TK protein kinase group
Tyrosine protein kinase Src family
|
- | - | - | - | 16 | |
|
Enzyme
Kinase
Protein Kinase
TK protein kinase group
Tyrosine protein kinase VEGFR family
|
- | 10-380 | 9.6-330 | - | 58 |
 Danio rerio
Danio rerio
 Homo sapiens
Homo sapiens
 Rattus norvegicus
Rattus norvegicus
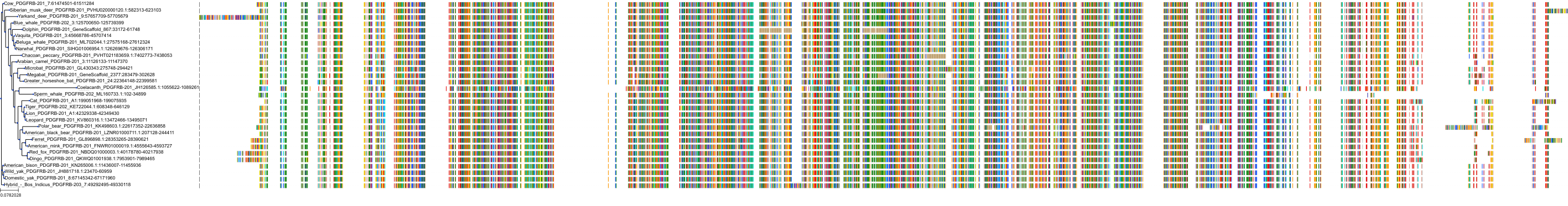
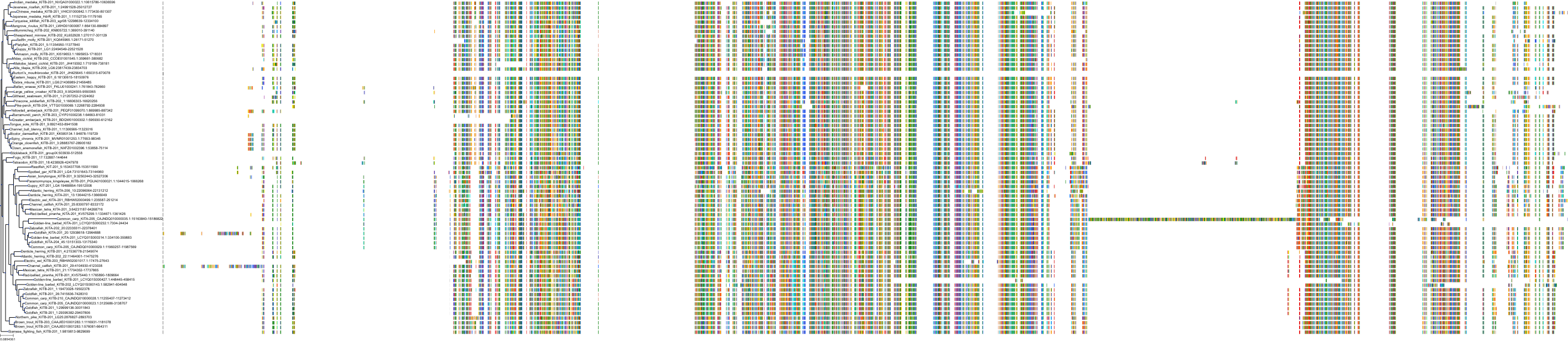
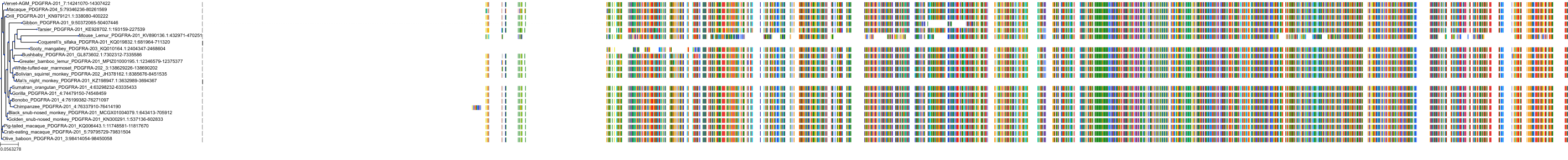
Target Conservation
|
Protein: Macrophage colony stimulating factor receptor Description: Macrophage colony-stimulating factor 1 receptor Organism : Homo sapiens P07333 ENSG00000182578 |
|
|||
|
Protein: Platelet-derived growth factor receptor Description: Platelet-derived growth factor receptor beta Organism : Homo sapiens P09619 ENSG00000113721 |
|
|||
|
Protein: Stem cell growth factor receptor Description: Mast/stem cell growth factor receptor Kit Organism : Homo sapiens P10721 ENSG00000157404 |
|
|||
|
Protein: Platelet-derived growth factor receptor Description: Platelet-derived growth factor receptor alpha Organism : Homo sapiens P16234 ENSG00000134853 |
|
|||
|
Protein: Vascular endothelial growth factor receptor Description: Vascular endothelial growth factor receptor 1 Organism : Homo sapiens P17948 ENSG00000102755 |
|
|||
|
Protein: Vascular endothelial growth factor receptor Description: Vascular endothelial growth factor receptor 3 Organism : Homo sapiens P35916 ENSG00000037280 |
|
|||
|
Protein: Vascular endothelial growth factor receptor Description: Vascular endothelial growth factor receptor 2 Organism : Homo sapiens P35968 ENSG00000128052 |
|
|||










































































Cross References
| Resources | Reference |
|---|---|
| CAS NUMBER | 212141-54-3 |
| ChEBI | 90620 |
| ChEMBL | CHEMBL101253 |
| DrugBank | DB04879 |
| FDA SRS | 5DX9U76296 |
| Guide to Pharmacology | 5705 |
| PubChem | 151194 |
| SureChEMBL | SCHEMBL18286 |
| ZINC | ZINC000000007460 |
CONTENTS
