
| Synonyms | |
| Status | |
| Molecule Category | Free-form |
| UNII | 3S9RX35S5X |
| EPA CompTox | DTXSID90712307 |
Structure
| InChI Key | JOWXJLIFIIOYMS-UHFFFAOYSA-N |
|---|---|
| Smiles | |
| InChI |
|
Physicochemical Descriptors
| Property Name | Value |
|---|---|
| Molecular Formula | C23H24N8O4S |
| Molecular Weight | 508.56 |
| AlogP | 2.14 |
| Hydrogen Bond Acceptor | 12.0 |
| Hydrogen Bond Donor | 2.0 |
| Number of Rotational Bond | 7.0 |
| Polar Surface Area | 138.72 |
| Molecular species | NEUTRAL |
| Aromatic Rings | 4.0 |
| Heavy Atoms | 36.0 |
Pharmacology
| Mechanism of Action | Action | Reference |
|---|---|---|
| Histone deacetylase inhibitor | INHIBITOR | PubMed Other |
| PI3-kinase p110-alpha subunit inhibitor | INHIBITOR | PubMed Other |
| PI3-kinase p110-beta subunit inhibitor | INHIBITOR | PubMed Other |
| PI3-kinase p110-delta subunit inhibitor | INHIBITOR | PubMed Other |
| Targets | EC50(nM) | IC50(nM) | Kd(nM) | Ki(nM) | Inhibition(%) | |
|---|---|---|---|---|---|---|
|
Enzyme
Kinase
Protein Kinase
Atypical protein kinase group
Atypical protein kinase PIKK family
Atypical protein kinase FRAP subfamily
|
- | 185-431 | - | - | - | |
|
Enzyme
Transferase
|
- | 6.3-374 | - | - | - | |
|
Enzyme
|
- | 6.3-374 | - | - | - | |
|
Epigenetic regulator
Eraser
Histone deacetylase
HDAC class I
|
126.25 | 0.36-426 | - | - | - | |
|
Epigenetic regulator
Eraser
Histone deacetylase
HDAC class IIa
|
- | 6.8-674 | - | - | - | |
|
Epigenetic regulator
Eraser
Histone deacetylase
HDAC class IIb
|
221.75 | 2.8-674 | - | - | - | |
|
Epigenetic regulator
Eraser
Histone deacetylase
HDAC class IV
|
- | 5-132 | - | - | - | |
|
Unclassified protein
|
- | 1.8-3.2 | - | - | - |
 Homo sapiens
Homo sapiens
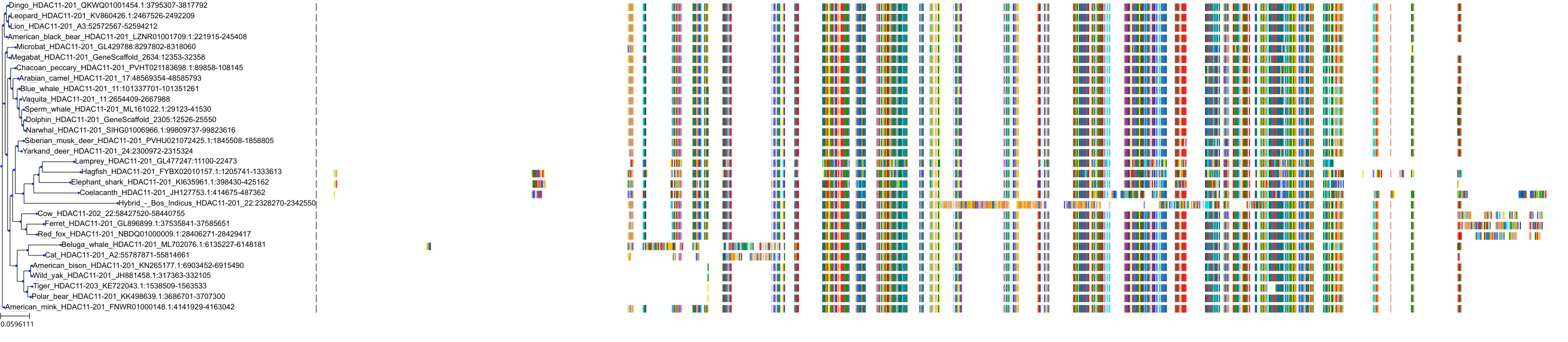
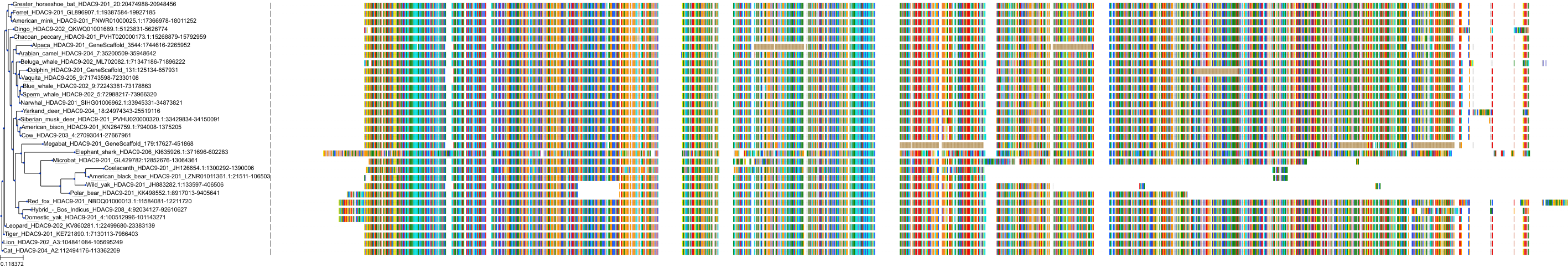
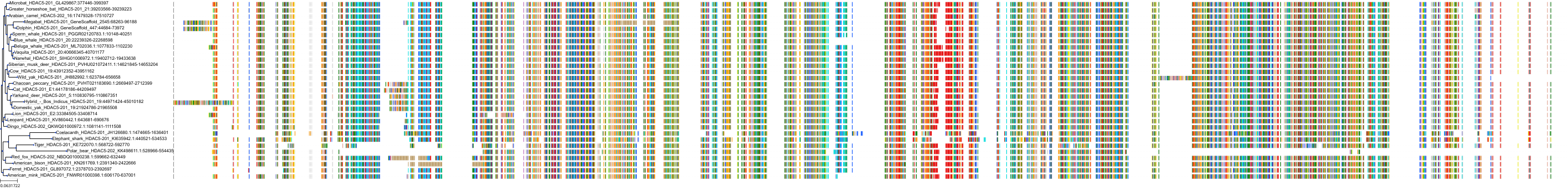
Target Conservation
|
Protein: PI3-kinase p110-delta subunit Description: Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit delta isoform Organism : Homo sapiens O00329 ENSG00000171608 |
|
|||
|
Protein: Histone deacetylase Description: Histone deacetylase 3 Organism : Homo sapiens O15379 ENSG00000171720 |
|
|||
|
Protein: PI3-kinase p110-alpha subunit Description: Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha isoform Organism : Homo sapiens P42336 ENSG00000121879 |
|
|||
|
Protein: PI3-kinase p110-beta subunit Description: Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit beta isoform Organism : Homo sapiens P42338 ENSG00000051382 |
|
|||
|
Protein: Histone deacetylase Description: Histone deacetylase 1 Organism : Homo sapiens Q13547 ENSG00000116478 |
|
|||
|
Protein: Histone deacetylase Description: Histone deacetylase 7 Organism : Homo sapiens Q8WUI4 ENSG00000061273 |
|
|||
|
Protein: Histone deacetylase Description: Histone deacetylase 2 Organism : Homo sapiens Q92769 ENSG00000196591 |
|
|||
|
Protein: Histone deacetylase Description: Polyamine deacetylase HDAC10 Organism : Homo sapiens Q969S8 ENSG00000100429 |
|
|||
|
Protein: Histone deacetylase Description: Histone deacetylase 11 Organism : Homo sapiens Q96DB2 ENSG00000163517 |
|
|||
|
Protein: Histone deacetylase Description: Histone deacetylase 8 Organism : Homo sapiens Q9BY41 ENSG00000147099 |
|
|||
|
Protein: Histone deacetylase Description: Histone deacetylase 6 Organism : Homo sapiens Q9UBN7 ENSG00000094631 |
|
|||
|
Protein: Histone deacetylase Description: Histone deacetylase 9 Organism : Homo sapiens Q9UKV0 ENSG00000048052 |
|
|||
|
Protein: Histone deacetylase Description: Histone deacetylase 5 Organism : Homo sapiens Q9UQL6 ENSG00000108840 |
|
|||






































































































































Cross References
| Resources | Reference |
|---|---|
| CAS NUMBER | 1339928-25-4 |
| ChEMBL | CHEMBL3622533 |
| DrugBank | DB11891 |
| FDA SRS | 3S9RX35S5X |
| Guide to Pharmacology | 8952 |
| PubChem | 54575456 |
| SureChEMBL | SCHEMBL1284705 |
| ZINC | ZINC000073488511 |
CONTENTS
