
| Synonyms | |
| Status | |
| Molecule Category | Free-form |
| ATC | C04AX32 |
| UNII | Q0CH43PGXS |
| EPA CompTox | DTXSID4048569 |
Structure
| InChI Key | NGOGFTYYXHNFQH-UHFFFAOYSA-N |
|---|---|
| Smiles | |
| InChI |
|
Physicochemical Descriptors
| Property Name | Value |
|---|---|
| Molecular Formula | C14H17N3O2S |
| Molecular Weight | 291.38 |
| AlogP | 1.22 |
| Hydrogen Bond Acceptor | 4.0 |
| Hydrogen Bond Donor | 1.0 |
| Number of Rotational Bond | 2.0 |
| Polar Surface Area | 62.3 |
| Molecular species | NEUTRAL |
| Aromatic Rings | 2.0 |
| Heavy Atoms | 20.0 |
Pharmacology
| Mechanism of Action | Action | Reference |
|---|---|---|
| Rho-associated protein kinase inhibitor | INHIBITOR | PubMed |
 Bos taurus
Bos taurus
 Homo sapiens
Homo sapiens
 Oryctolagus cuniculus
Oryctolagus cuniculus
 Ovis aries
Ovis aries
 Rattus norvegicus
Rattus norvegicus
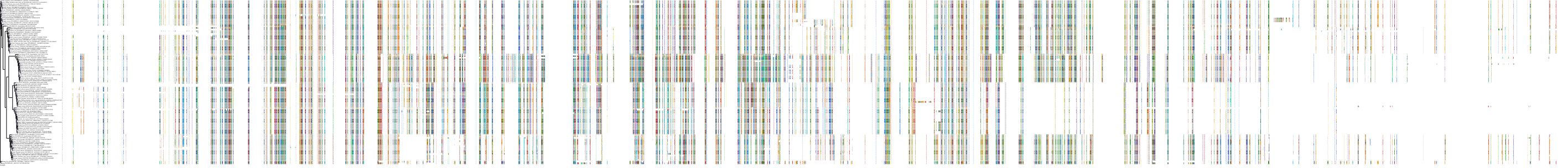
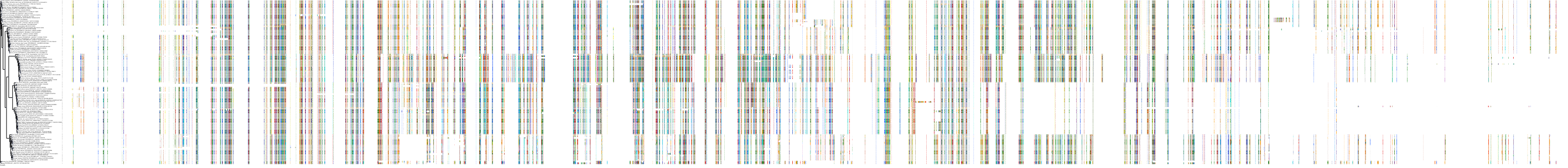
Target Conservation
|
Protein: Rho-associated protein kinase Description: Rho-associated protein kinase 2 Organism : Homo sapiens O75116 ENSG00000134318 |
|
|||
|
Protein: Rho-associated protein kinase Description: Rho-associated protein kinase 1 Organism : Homo sapiens Q13464 ENSG00000067900 |
|
|||




















Related Entries

Cross References
| Resources | Reference |
|---|---|
| CAS NUMBER | 103745-39-7 |
| ChEMBL | CHEMBL38380 |
| DrugBank | DB08162 |
| DrugCentral | 1132 |
| FDA SRS | Q0CH43PGXS |
| Guide to Pharmacology | 5181 |
| PDB | M77 |
| PubChem | 3547 |
| SureChEMBL | SCHEMBL4674 |
| ZINC | ZINC000000006486 |
CONTENTS
