
| Synonyms | |
| Status | |
| Molecule Category | Free-form |
| UNII | WFR23M21IE |
| EPA CompTox | DTXSID40150710 |
Structure
| InChI Key | JDUBGYFRJFOXQC-KRWDZBQOSA-N |
|---|---|
| Smiles | |
| InChI |
|
Physicochemical Descriptors
| Property Name | Value |
|---|---|
| Molecular Formula | C21H25ClN6O2 |
| Molecular Weight | 428.92 |
| AlogP | 2.15 |
| Hydrogen Bond Acceptor | 6.0 |
| Hydrogen Bond Donor | 4.0 |
| Number of Rotational Bond | 6.0 |
| Polar Surface Area | 120.16 |
| Molecular species | NEUTRAL |
| Aromatic Rings | 3.0 |
| Heavy Atoms | 30.0 |
Pharmacology
| Mechanism of Action | Action | Reference |
|---|---|---|
| Serine/threonine-protein kinase AKT inhibitor | INHIBITOR |
| Targets | EC50(nM) | IC50(nM) | Kd(nM) | Ki(nM) | Inhibition(%) | |
|---|---|---|---|---|---|---|
|
Enzyme
Kinase
Protein Kinase
AGC protein kinase group
AGC protein kinase AKT family
|
- | 3-34 | - | - | 85-99 | |
|
Enzyme
Kinase
Protein Kinase
AGC protein kinase group
AGC protein kinase DMPK family
AGC protein kinase ROCK subfamily
|
- | 56-470 | - | - | - | |
|
Ion channel
Voltage-gated ion channel
Potassium channels
Voltage-gated potassium channel
|
- | - | - | - | 7.1 |
 Homo sapiens
Homo sapiens
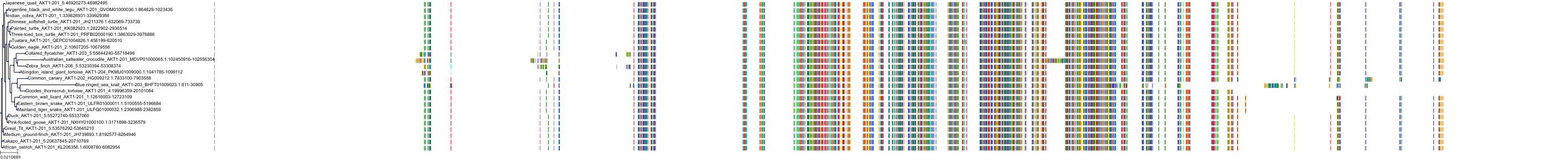
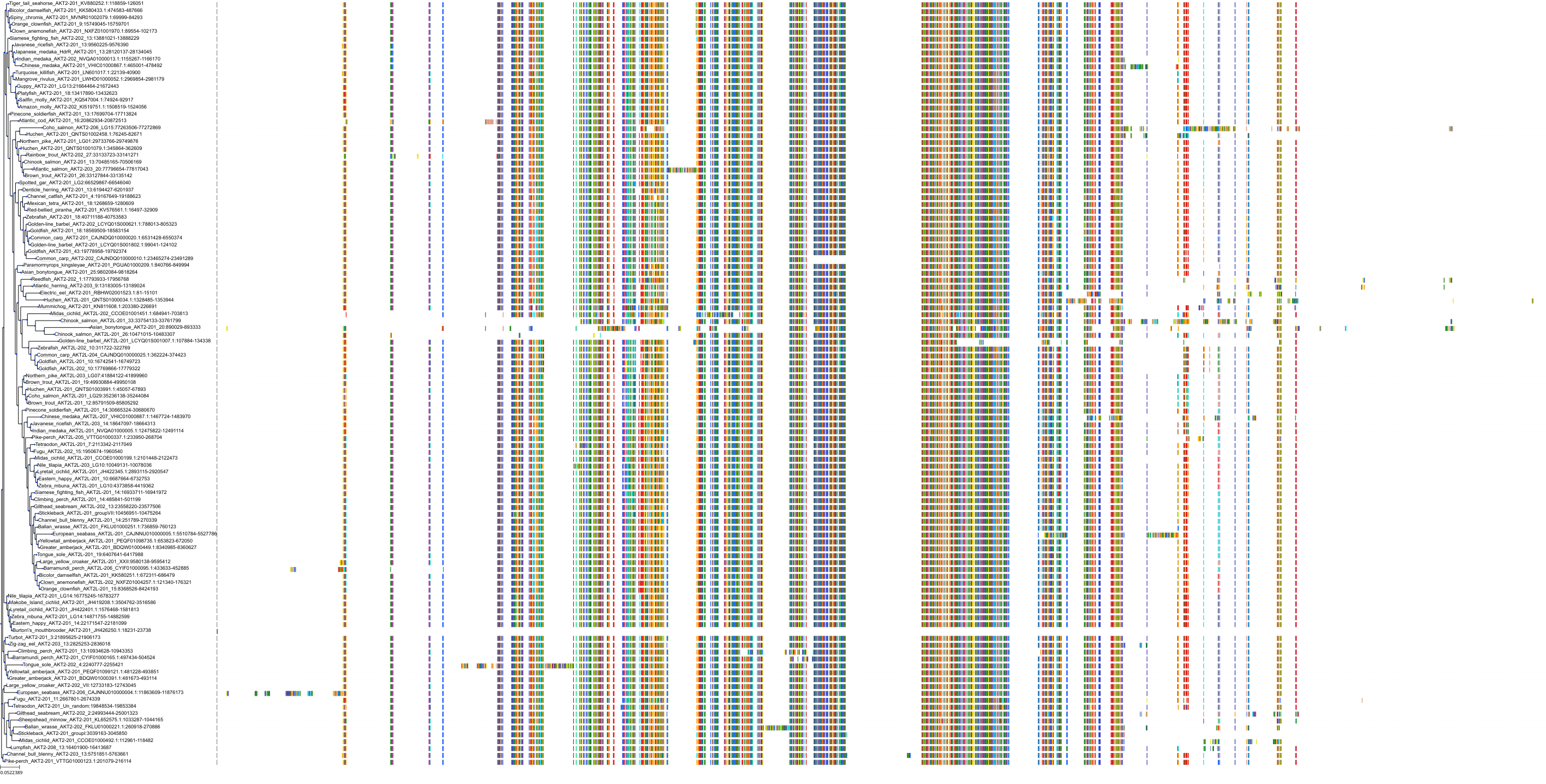
Target Conservation
|
Protein: Serine/threonine-protein kinase AKT Description: RAC-alpha serine/threonine-protein kinase Organism : Homo sapiens P31749 ENSG00000142208 |
|
|||
|
Protein: Serine/threonine-protein kinase AKT Description: RAC-beta serine/threonine-protein kinase Organism : Homo sapiens P31751 ENSG00000105221 |
|
|||
|
Protein: Serine/threonine-protein kinase AKT Description: RAC-gamma serine/threonine-protein kinase Organism : Homo sapiens Q9Y243 ENSG00000117020 |
|
|||





























Cross References
| Resources | Reference |
|---|---|
| CAS NUMBER | 1143532-39-1 |
| ChEMBL | CHEMBL2325741 |
| DrugBank | DB12218 |
| FDA SRS | WFR23M21IE |
| Guide to Pharmacology | 7709 |
| PDB | 0XZ |
| PubChem | 25227436 |
| SureChEMBL | SCHEMBL390243 |
| ZINC | ZINC000043204023 |
CONTENTS
