
| Synonyms | |
| Status | |
| Molecule Category | Free-form |
| UNII | 001L2FE0M3 |
| EPA CompTox | DTXSID00963646 |
Structure
| InChI Key | KUFRQPKVAWMTJO-LMZWQJSESA-N |
|---|---|
| Smiles | |
| InChI |
|
Physicochemical Descriptors
| Property Name | Value |
|---|---|
| Molecular Formula | C32H48N4O8 |
| Molecular Weight | 616.76 |
| AlogP | 1.91 |
| Hydrogen Bond Acceptor | 10.0 |
| Hydrogen Bond Donor | 4.0 |
| Number of Rotational Bond | 7.0 |
| Polar Surface Area | 169.52 |
| Molecular species | NEUTRAL |
| Aromatic Rings | 0.0 |
| Heavy Atoms | 44.0 |
Pharmacology
| Mechanism of Action | Action | Reference |
|---|---|---|
| Heat shock protein HSP90 inhibitor | INHIBITOR | PubMed |
| Reactive oxygen species | None | PubMed |
| Targets | EC50(nM) | IC50(nM) | Kd(nM) | Ki(nM) | Inhibition(%) | |
|---|---|---|---|---|---|---|
|
Other cytosolic protein
|
4-62 | 4.5-930 | 87-500 | 680 | 88.31 | |
|
Other membrane protein
|
65 | - | - | - | - | |
|
Transcription factor
|
- | 3.6-61 | - | - | - |
 Canis lupus familiaris
Canis lupus familiaris
 Hepatitis C virus
Hepatitis C virus
 Homo sapiens
Homo sapiens
 Mus musculus
Mus musculus
Target Conservation
|
Protein: Heat shock protein HSP90 Description: Heat shock protein HSP 90-alpha Organism : Homo sapiens P07900 ENSG00000080824 |
|
|||
|
Protein: Heat shock protein HSP90 Description: Heat shock protein HSP 90-beta Organism : Homo sapiens P08238 ENSG00000096384 |
|
|||



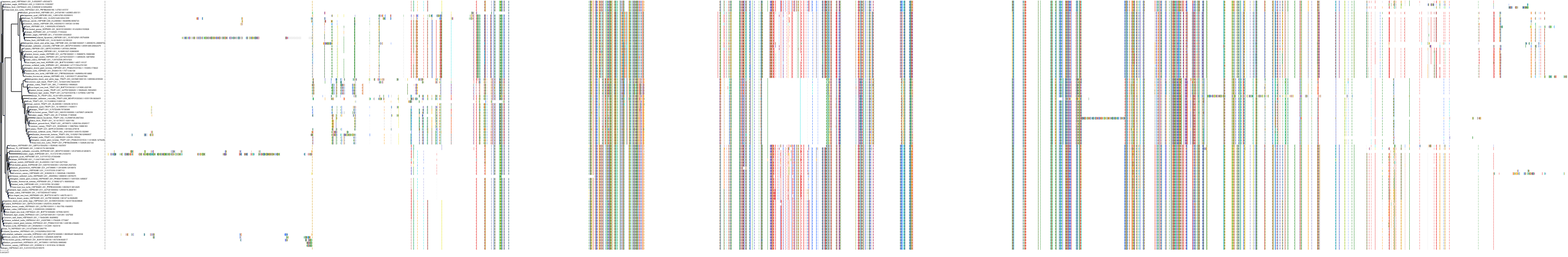










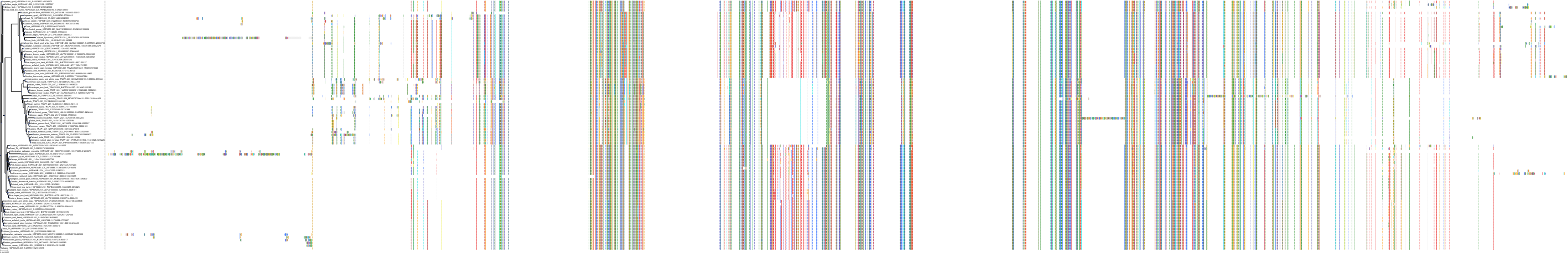





Related Entries

Cross References
| Resources | Reference |
|---|---|
| CAS NUMBER | 467214-20-6 |
| ChEBI | 65324 |
| ChEMBL | CHEMBL383824 |
| DrugBank | DB12442 |
| FDA SRS | 001L2FE0M3 |
| Guide to Pharmacology | 9828 |
| PDB | KOS |
| PubChem | 5288674 |
| SureChEMBL | SCHEMBL5449716 |
| ZINC | ZINC000100030312 |
CONTENTS
