
| Trade Names | |
| Synonyms | |
| Status | |
| Molecule Category | Free-form |
| ATC | L01EC01 |
| UNII | 207SMY3FQT |
| EPA CompTox | DTXSID50238710 |
Structure
| InChI Key | GPXBXXGIAQBQNI-UHFFFAOYSA-N |
|---|---|
| Smiles | |
| InChI |
|
Physicochemical Descriptors
| Property Name | Value |
|---|---|
| Molecular Formula | C23H18ClF2N3O3S |
| Molecular Weight | 489.93 |
| AlogP | 5.54 |
| Hydrogen Bond Acceptor | 4.0 |
| Hydrogen Bond Donor | 2.0 |
| Number of Rotational Bond | 7.0 |
| Polar Surface Area | 91.92 |
| Molecular species | NEUTRAL |
| Aromatic Rings | 4.0 |
| Heavy Atoms | 33.0 |
Pharmacology
| Mechanism of Action | Action | Reference |
|---|---|---|
| Serine/threonine-protein kinase B-raf inhibitor | INHIBITOR | FDA |
| Targets | EC50(nM) | IC50(nM) | Kd(nM) | Ki(nM) | Inhibition(%) | |
|---|---|---|---|---|---|---|
|
Enzyme
Kinase
Protein Kinase
STE protein kinase group
STE protein kinase STE7 family
|
- | - | 8.2 | - | - | |
|
Enzyme
Kinase
Protein Kinase
TK protein kinase group
Tyrosine protein kinase VEGFR family
|
- | 360 | - | - | - | |
|
Enzyme
Kinase
Protein Kinase
TKL protein kinase group
TKL protein kinase MLK family
TKL protein kinase MLK subfamily
|
- | 23-31.4 | - | - | - | |
|
Enzyme
Kinase
Protein Kinase
TKL protein kinase group
TKL protein kinase RAF family
|
601-794.33 | 2.1-950 | 50.7-58.2 | - | 72-95 | |
|
Enzyme
Oxidoreductase
|
- | - | - | - | 81.1 |
 Homo sapiens
Homo sapiens
 Mus musculus
Mus musculus
Target Conservation
|
Protein: Serine/threonine-protein kinase B-raf Description: Serine/threonine-protein kinase B-raf Organism : Homo sapiens P15056 ENSG00000157764 |
|
|||









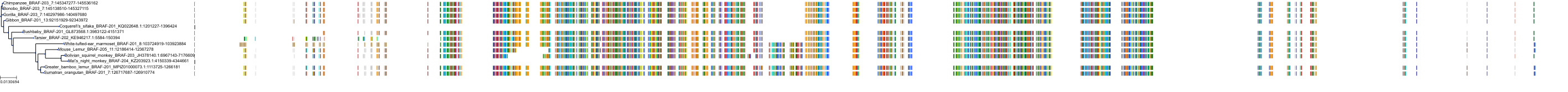

Cross References
| Resources | Reference |
|---|---|
| CAS NUMBER | 918504-65-1 |
| ChEBI | 63637 |
| ChEMBL | CHEMBL1229517 |
| DrugBank | DB08881 |
| DrugCentral | 4185 |
| FDA SRS | 207SMY3FQT |
| Guide to Pharmacology | 5893 |
| KEGG | D09996 |
| PDB | 032 |
| PharmGKB | PA165946873 |
| PubChem | 42611257 |
| SureChEMBL | SCHEMBL298931 |
| ZINC | ZINC000052509366 |
CONTENTS
