
| Trade Names | |
| Synonyms | |
| Status | |
| Molecule Category | Free-form |
| ATC | L01EM01 |
| UNII | YG57I8T5M0 |
| EPA CompTox | DTXSID701007266 |
Structure
| InChI Key | IFSDAJWBUCMOAH-HNNXBMFYSA-N |
|---|---|
| Smiles | |
| InChI |
|
Physicochemical Descriptors
| Property Name | Value |
|---|---|
| Molecular Formula | C22H18FN7O |
| Molecular Weight | 415.43 |
| AlogP | 3.75 |
| Hydrogen Bond Acceptor | 7.0 |
| Hydrogen Bond Donor | 2.0 |
| Number of Rotational Bond | 5.0 |
| Polar Surface Area | 101.38 |
| Molecular species | NEUTRAL |
| Aromatic Rings | 5.0 |
| Heavy Atoms | 31.0 |
Pharmacology
| Mechanism of Action | Action | Reference |
|---|---|---|
| PI3-kinase p110-delta subunit inhibitor | INHIBITOR | FDA |
| Targets | EC50(nM) | IC50(nM) | Kd(nM) | Ki(nM) | Inhibition(%) | |
|---|---|---|---|---|---|---|
|
Enzyme
Transferase
|
- | 1.08-980 | - | - | 91.91-96 | |
|
Enzyme
|
- | 1.08-980 | - | - | 91.91-96 |
 Homo sapiens
Homo sapiens
 Rattus norvegicus
Rattus norvegicus
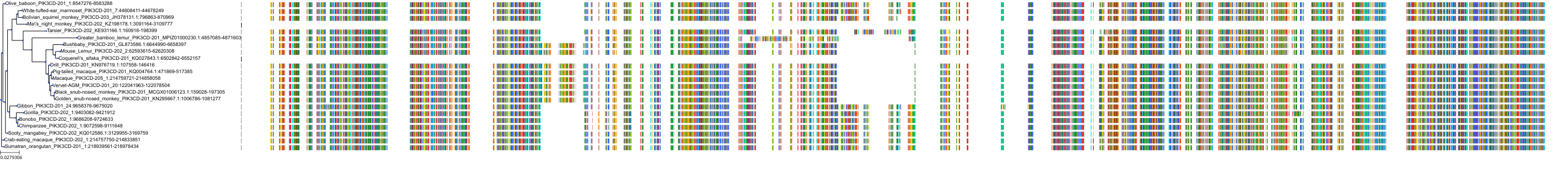
Target Conservation
|
Protein: PI3-kinase p110-delta subunit Description: Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit delta isoform Organism : Homo sapiens O00329 ENSG00000171608 |
|
|||










Related Entries

Cross References
| Resources | Reference |
|---|---|
| CAS NUMBER | 870281-82-6 |
| ChEBI | 82701 |
| ChEMBL | CHEMBL2216870 |
| DrugBank | DB09054 |
| DrugCentral | 4878 |
| FDA SRS | YG57I8T5M0 |
| Guide to Pharmacology | 6741 |
| PDB | 40L |
| PubChem | 11625818 |
| SureChEMBL | SCHEMBL356400 |
| ZINC | ZINC000013986658 |
CONTENTS
