
| Trade Names | |
| Synonyms | |
| Status | |
| Molecule Category | Free-form |
| ATC | N01AH01 N02AB03 |
| UNII | UF599785JZ |
| EPA CompTox | DTXSID9023049 |
Structure
| InChI Key | PJMPHNIQZUBGLI-UHFFFAOYSA-N |
|---|---|
| Smiles | |
| InChI |
|
Physicochemical Descriptors
| Property Name | Value |
|---|---|
| Molecular Formula | C22H28N2O |
| Molecular Weight | 336.48 |
| AlogP | 4.14 |
| Hydrogen Bond Acceptor | 2.0 |
| Number of Rotational Bond | 6.0 |
| Polar Surface Area | 23.55 |
| Molecular species | BASE |
| Aromatic Rings | 2.0 |
| Heavy Atoms | 25.0 |
Pharmacology
| Mechanism of Action | Action | Reference |
|---|---|---|
| Mu opioid receptor agonist | AGONIST | DailyMed |
| Targets | EC50(nM) | IC50(nM) | Kd(nM) | Ki(nM) | Inhibition(%) | |
|---|---|---|---|---|---|---|
|
Membrane receptor
Family A G protein-coupled receptor
Peptide receptor (family A GPCR)
Short peptide receptor (family A GPCR)
Opioid receptor
|
0.51-53 | 1.14-431 | - | 1.1-679 | - | |
|
Membrane receptor
Family A G protein-coupled receptor
Small molecule receptor (family A GPCR)
Monoamine receptor
Dopamine receptor
|
- | - | - | 554 | - | |
|
Membrane receptor
|
0.51-53 | 1.14-431 | - | 1.1-679 | - | |
|
Transporter
Electrochemical transporter
SLC superfamily of solute carriers
SLC21/SLCO family of organic anion transporting polypeptides
|
- | - | - | - | -53.3-22.6 | |
|
Transporter
Electrochemical transporter
SLC superfamily of solute carriers
SLC22 family of organic cation and anion transporters
|
- | - | - | - | 73.6 |
 Cavia porcellus
Cavia porcellus
 Homo sapiens
Homo sapiens
 Mus musculus
Mus musculus
 Rattus norvegicus
Rattus norvegicus
Target Conservation
|
Protein: Mu opioid receptor Description: Mu-type opioid receptor Organism : Homo sapiens P35372 ENSG00000112038 |
|
|||



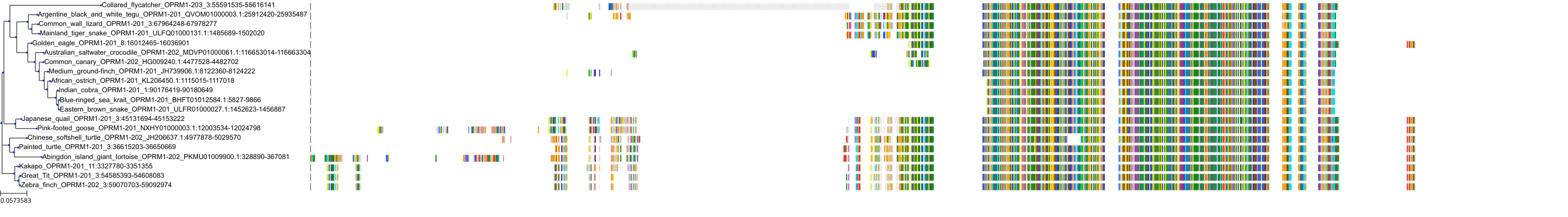




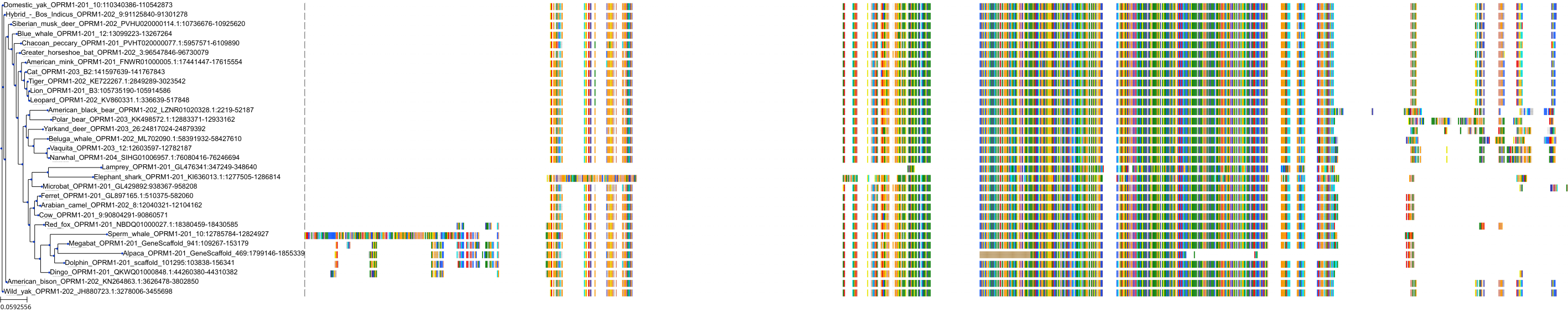


Environmental Exposure
Cross References
| Resources | Reference |
|---|---|
| CAS NUMBER | 437-38-7 |
| ChEBI | 119915 |
| ChEMBL | CHEMBL596 |
| DrugBank | DB00813 |
| DrugCentral | 1164 |
| FDA SRS | UF599785JZ |
| Human Metabolome Database | HMDB0014951 |
| Guide to Pharmacology | 1626 |
| PDB | 7V7 |
| PharmGKB | PA449599 |
| PubChem | 3345 |
| SureChEMBL | SCHEMBL8804 |
| ZINC | ZINC000002522669 |
CONTENTS
