
| Trade Names | |
| Synonyms | |
| Status | |
| Molecule Category | Free-form |
| ATC | M04AA01 |
| UNII | 63CZ7GJN5I |
| EPA CompTox | DTXSID4022573 |
Structure
| InChI Key | OFCNXPDARWKPPY-UHFFFAOYSA-N |
|---|---|
| Smiles | |
| InChI |
|
Physicochemical Descriptors
| Property Name | Value |
|---|---|
| Molecular Formula | C5H4N4O |
| Molecular Weight | 136.11 |
| AlogP | -0.4 |
| Hydrogen Bond Acceptor | 3.0 |
| Hydrogen Bond Donor | 2.0 |
| Polar Surface Area | 74.43 |
| Molecular species | NEUTRAL |
| Aromatic Rings | 0.0 |
| Heavy Atoms | 10.0 |
Pharmacology
| Mechanism of Action | Action | Reference |
|---|---|---|
| Xanthine dehydrogenase inhibitor | INHIBITOR | FDA |
| Targets | EC50(nM) | IC50(nM) | Kd(nM) | Ki(nM) | Inhibition(%) | |
|---|---|---|---|---|---|---|
|
Enzyme
Hydrolase
|
- | - | - | - | 76 | |
|
Enzyme
Oxidoreductase
|
- | 3.6-950 | 0.54 | 100-700 | 4-100 | |
|
Secreted protein
|
- | - | - | - | 76 | |
|
Transporter
Electrochemical transporter
SLC superfamily of solute carriers
SLC21/SLCO family of organic anion transporting polypeptides
|
- | - | - | - | -35.6-84.36 | |
|
Transporter
Electrochemical transporter
SLC superfamily of solute carriers
SLC22 family of organic cation and anion transporters
|
- | - | - | - | 7.7 |
 Agaricus bisporus
Agaricus bisporus
 Bos taurus
Bos taurus
 Cricetulus griseus
Cricetulus griseus
 Homo sapiens
Homo sapiens
 Mus musculus
Mus musculus
 Oryctolagus cuniculus
Oryctolagus cuniculus
 Rattus norvegicus
Rattus norvegicus
 Shigella dysenteriae
Shigella dysenteriae
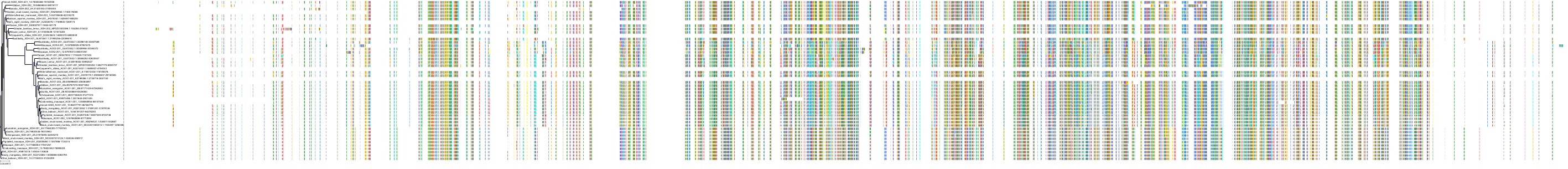
Target Conservation
|
Protein: Xanthine dehydrogenase Description: Xanthine dehydrogenase/oxidase Organism : Homo sapiens P47989 ENSG00000158125 |
|
|||










Cross References
| Resources | Reference |
|---|---|
| CAS NUMBER | 315-30-0 |
| ChEBI | 40279 |
| ChEMBL | CHEMBL1467 |
| DrugBank | DB00437 |
| DrugCentral | 124 |
| FDA SRS | 63CZ7GJN5I |
| Human Metabolome Database | HMDB0014581 |
| Guide to Pharmacology | 6795 |
| PharmGKB | PA448320 |
| PubChem | 135401907 |
| SureChEMBL | SCHEMBL4627 |
| ZINC | ZINC000013298313 |
CONTENTS
