
| Trade Names | |
| Synonyms | |
| Status | |
| Molecule Category | Free-form |
| ATC | L01BC09 |
| UNII | 039LU44I5M |
| EPA CompTox | DTXSID3023057 |
Structure
| InChI Key | ODKNJVUHOIMIIZ-RRKCRQDMSA-N |
|---|---|
| Smiles | |
| InChI |
|
Physicochemical Descriptors
| Property Name | Value |
|---|---|
| Molecular Formula | C9H11FN2O5 |
| Molecular Weight | 246.19 |
| AlogP | -1.68 |
| Hydrogen Bond Acceptor | 6.0 |
| Hydrogen Bond Donor | 3.0 |
| Number of Rotational Bond | 2.0 |
| Polar Surface Area | 104.55 |
| Molecular species | NEUTRAL |
| Aromatic Rings | 1.0 |
| Heavy Atoms | 17.0 |
Metabolites Network
Pharmacology
| Mechanism of Action | Action | Reference |
|---|---|---|
| Thymidylate synthase inhibitor | INHIBITOR | PubMed PubMed PubMed PubMed |
| Targets | EC50(nM) | IC50(nM) | Kd(nM) | Ki(nM) | Inhibition(%) | |
|---|---|---|---|---|---|---|
|
Enzyme
Transferase
|
- | 0.4-260 | - | - | - | |
|
Transporter
Electrochemical transporter
SLC superfamily of solute carriers
SLC21/SLCO family of organic anion transporting polypeptides
|
- | - | - | - | 109.16-116.26 | |
|
Transporter
Electrochemical transporter
SLC superfamily of solute carriers
SLC28 and SLC29 families of nucleoside transporters
SLC28 Na+-coupled nucleoside transport family
|
- | - | - | - | 17 |
 Chlorocebus sabaeus
Chlorocebus sabaeus
 Cricetulus griseus
Cricetulus griseus
 Cryptosporidium parvum
Cryptosporidium parvum
 Enterococcus faecium
Enterococcus faecium
.png) Herpes simplex virus (type 1 / strain CL101)
Herpes simplex virus (type 1 / strain CL101)
 Homo sapiens
Homo sapiens
 Human herpesvirus 1
Human herpesvirus 1
 Mus musculus
Mus musculus
 Toxoplasma gondii
Toxoplasma gondii
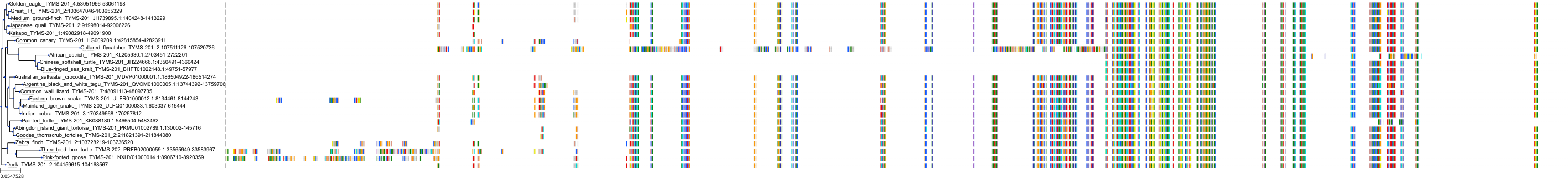
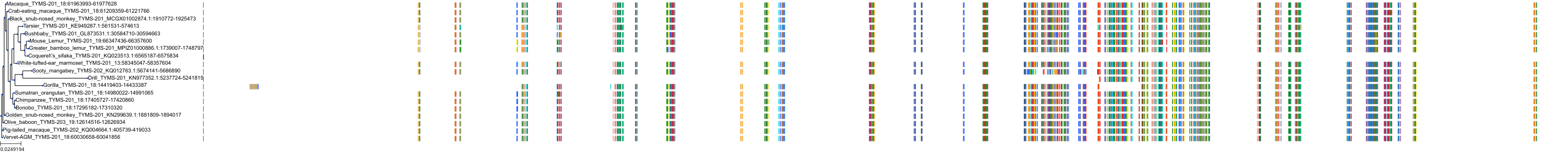
Target Conservation
|
Protein: Thymidylate synthase Description: Thymidylate synthase Organism : Homo sapiens P04818 ENSG00000176890 |
|
|||






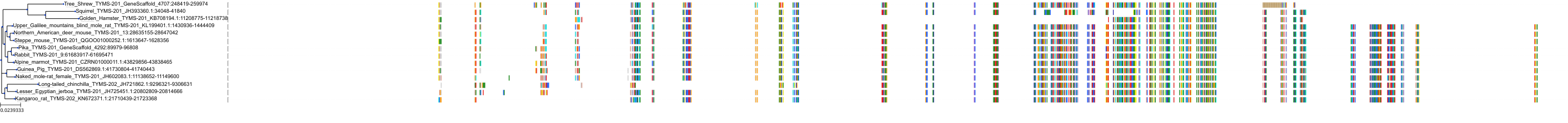


Related Entries

Cross References
| Resources | Reference |
|---|---|
| CAS NUMBER | 50-91-9 |
| ChEBI | 60761 |
| ChEMBL | CHEMBL917 |
| DrugBank | DB00322 |
| DrugCentral | 1184 |
| FDA SRS | 039LU44I5M |
| Human Metabolome Database | HMDB0014467 |
| Guide to Pharmacology | 4801 |
| KEGG | C11736 |
| PubChem | 5790 |
| SureChEMBL | SCHEMBL4424 |
| ZINC | ZINC000003813010 |
CONTENTS
